Eradigm Consulting
Fora, 180 Borough High Street,
London SE1 1L
Fora, 180 Borough High Street,
London SE1 1L
April 14 2023
Eradigm highlights the importance of robust evidence generation, optimised outcome-based agreements (OBAs), and impactful real-world evidence (RWE) for pharmaceutical companies to successfully navigate Europe's cost-saving environment during launch strategy planning.
While healthcare costs in Europe have been steadily rising since the turn of the millennium, the current inflationary macroeconomic environment has exacerbated the problem. Consequently, regulators across the EU have drafted legislation to reign in these costs before national healthcare deficits spiral out of control. In the UK, draft legislation threatens to increase the voluntary scheme for branded medicines pricing and access (VPAS) rebates from 24.4% to 27.5% to mitigate the budget deficit,1 while Germany seeks to tighten the among procedure to increase rebates.2 This is a zero-sum paradigm, whereby decreasing the healthcare deficit takes revenue away from pharmaceutical companies and reduces the industry’s ability to innovate. With continued cost-containment efforts, the pricing and reimbursement environment becomes even more challenging.
To ensure successful market access in Europe’s cost-saving environment, we propose redefining the approach to reimbursement, replacing steep discounts and rebates with evidence-based pricing, and encouraging manufacturers and payers to co-create mutually beneficial OBAs. This will begin with a foundation of appropriate evidence generation, which can support negotiation of optimised OBAs that are then supported by robust RWE practices (Figure 1).
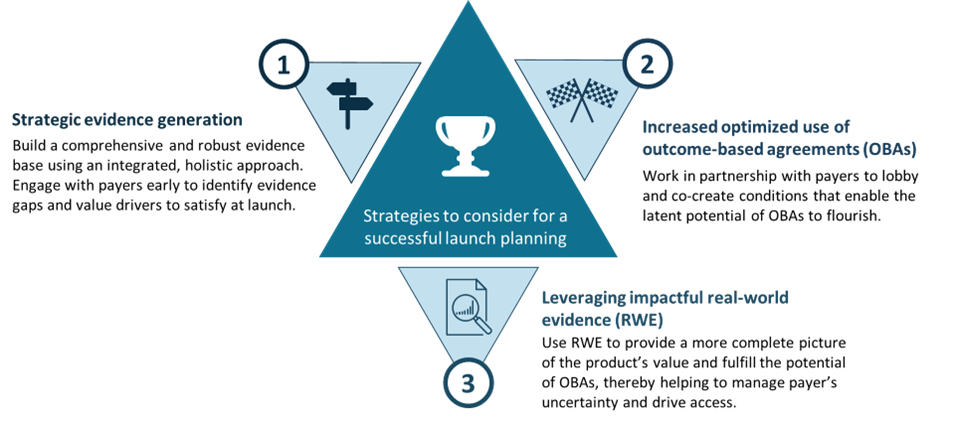
Achieving launch success begins with building a comprehensive and robust evidence base to support market access and pricing strategies.
The growth of HTA has spurred the need for evidence beyond regulatory data to inform reimbursement and decision-making. Payers generally require evidence of superior efficacy or tolerability, patient-relevant outcomes (including health-related quality of life), and health economics data to justify reimbursement and access to new products.3 Yet, payers’ specifications regarding acceptable comparators, endpoints, desirable thresholds, and types of data are often stringent and variable across markets.3 These evidence expectations, along with cost-containment efforts, have a significant impact on launch strategies and market access outcomes. This is illustrated in the market withdrawals of Tecvayli (teclistamab) in 2022 and Opdualag (nivolumab and relatlimab) in early 2023 in Germany. Since Germany introduced the “Financial Stabilisation of the Statutory Health Insurance System” (GKV-Finanzstabilisierungsgesetz) bill, it is crucial for manufacturers to prove additional benefit over existing treatments for price negotiations.4 However, despite Tecvayli receiving conditional EMA approval based on phase 1/2 evidence and Opdualag demonstrating statistically significant PFS, both products would expect a negative HTA outcome in Germany as the current AMNOG system does not recognise the added value of the therapies with the currently available evidence.5,6 Moreover, HTA bodies are periodically updating their methodological guidance, raising hurdles to generating robust evidence. For example, in 2020, the German HTA organisation (Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen, IQWiG) stipulated a 15% response threshold for all patient-reported outcomes for which an added benefit rating can be determined for a new intervention.7 Challenges thus remain in developing evidence that satisfies both regulatory and HTA agencies’ requirements across Europe at launch. Manufacturers should utilise early scientific advice channels and develop a clear market prioritisation plan to ensure the evidence can be tailored to match the needs of the market with the highest specification to entry.
The launch strategy in Europe is divided across markets and payer archetypes, presenting varied evidence requirements and market access challenges for manufacturers. Some markets, such as the UK, Sweden, and the Netherlands, focus more heavily on cost-effectiveness, while others, namely France and Germany, are driven by clinical evidence standards. While the payer archetype approach allows manufacturers to outline similar evidence requirements to leverage in the planning of launch and access strategies in Europe, the payer landscape is evolving. Markets are not necessarily fixed into one ‘archetype’ and may have room for variation in terms of payer value drivers, with increasing focus on cost justification. For example, Italy and Spain typically prioritised the overall budget impact but have increasingly incorporated cost-effectiveness analyses into formal consideration.8,9 In Spain, a high budget impact and/or lack of cost-effectiveness normally result in negative funding decisions, as shown in the therapeutic positioning reports (IPTs) of Talzenna (talazoparib) and Tecartus (brexucabtagene autoleucel).10,11 Meanwhile, in Italy, a recent study highlighted cost-effectiveness analyses with an ICER of ≥ €40,000/QALY had a significant impact on price negotiations resulting in approximately 30% price reduction, particularly for non-onco-immunological drugs and drugs for non-rare diseases.12 Across Europe, manufacturers also face multiple local, regional, and national decision-makers. Each of these bodies has its own priorities for cost-savings and improving patient outcomes, adding a further layer of complexity for launch considerations. A deep understanding of who the payer is and the key drivers influencing their decision-making will be required to inform both the evidence generation planning and launch strategy.
The evidence generation process has become increasingly difficult as the global market access landscape is evolving. Managing the needs and expectations of a broad range of payers, who are becoming more budget conscious, is key to launch success. Manufacturers will need to transition to a more holistic, integrated evidence-generation planning approach to prove the value of their products to various payers across the product lifecycle. This will require cross-functional collaboration across clinical development, medical affairs, market access, and HEOR teams on establishing strategic objectives, identifying evidence gaps, designing and executing studies, and building a communication strategy to convey the outcomes. Through this integrated approach, opportunities can be leveraged by anticipating and prioritizing all evidence gaps and determining overlapping evidence requirements across different payers. The integrated evidence plan should be regularly updated to reflect the latest changes in the asset strategy and evidence requirements.
In addition, manufacturers must engage with payers much earlier in the product development cycle to optimise the trial design or generate additional evidence to satisfy specific market access requirements. This includes selecting the appropriate comparator, targeting the correct patient population, investigating the most compelling and relevant endpoint, and the use of indirect treatment comparison and RWE. Early and ongoing engagement with payers will provide important insights into their needs and expectations that can later help shape potential value propositions, market access and pricing strategies for launch.
Innovative payment models, collectively termed managed entry agreements (MEAs), offer an alternative to the traditional list price discount and can increase the likelihood of reimbursement, but only if tailored appropriately to the market.
MEAs can be broadly divided into two buckets: financial and performance based. To date, financial MEAs have been the prevailing model due to their relative ease of implementation, and account for 79% of all MEAs.13 The key to their success is operational simplicity, functioning to control costs to the healthcare system. Price volume agreements (PVAs) is a commonly used example of financial MEAs, in which the unit price is linked to volume sold, enabling expenditure to be controlled if the product is approved in a wider patient population.14 The UK has been a leading adopter of financial MEAs, leveraging patient access schemes (PAS) as an access route for innovative products that improves cost-effectiveness by seeking commitments to rebates and discounts from manufacturers.15 While the implications of this exit for patient access are still emerging, it is clear that the financial MEA is not appropriate in the current macroeconomic environment, as the rebates needed to cover increasing deficits do not support research and commercialisation. Therefore, alternative reimbursement models must be utilised to find a compromise that is mutually beneficial to payers and manufacturers.
The second type of MEA, performance-based agreements, may provide an answer. Often termed outcome-based agreements (OBAs), they seek to link real-world treatment outcomes to variable payments. However, such models have never reached the same heights of popularity as financial MEAs due to their implementation complexity; to be effective, the logistical and administrative costs of the OBA must be lower than the savings it produces.16 If optimally designed, OBAs de-risk reimbursement by spreading the cost of a product over multiple installments and ensuring that the payer only pays for the patient outcomes observed. Such contracts will become increasingly important in the era of cell and gene therapies, where manufacturers set a high list price to account for their ‘curative’ potential, but which represents a greater risk to payers due to the limited evidence available. Payers across Europe have already demonstrated a strong appetite for OBAs in response to this, with Italy and Spain paving the way for the first generation of CD-19 CAR-Ts, such as Yescarta and Kymriah. These contracts went a step further to combine financial MEAs with OBAs to secure an initial discount to list price, followed up by results-linked payments, providing a clear example of positive payer-manufacturer interaction.17
Despite payers in other EU markets expressing a need for OBAs to cope with high costs, there remain structural barriers blocking the way. In France, the Economic Committee for Health Products (CEPS) has highlighted that simplicity makes straight discounts the preferred method of cost control, with OBAs instead seen as a tool to account for the discrepancy in real-world effectiveness.18 The underlying reason for this is that the French healthcare system operates on a 1-year budget cycle, leaving payers unable to account for lifetime cost savings during the HTA process. Consequently, high price curative therapies are encountering difficulties launching in the French market, and payers are left without the right tools to provide patient access. Across the border in Germany, where legislators introduced a cost containment bill for 2023 aiming to reduce the nation’s €17B healthcare deficit,19 payers are calling for administrative simplicity to make OBAs cheaper and easier to implement.
In both cases, manufacturers must work in partnership with payers to lobby and co-create conditions that enable the latent potential of OBAs to flourish. To ensure an effective launch, this must be started at an early stage, alongside clinical trial design, to establish synergy between the launch conditions and product profile. However, pre-launch work is insufficient by itself; to seal the deal, the generation, analysis, and utilisation of RWE must be designed to suit individual market demands while minimising the administrative burden.
As pressure mounts to contain costs, payers are increasingly forced to restrict access to drugs, often citing the lack of clear and compelling effectiveness data as the rationale. However, the use of RWE has transformed healthcare by providing a more complete picture of the product’s value in the ‘real world’, thereby helping to manage payers’ uncertainty and facilitate access. Harnessing the power of RWE for launch and market access will require a better understanding of how it should be collected, analysed, and ultimately utilised (Figure 2).
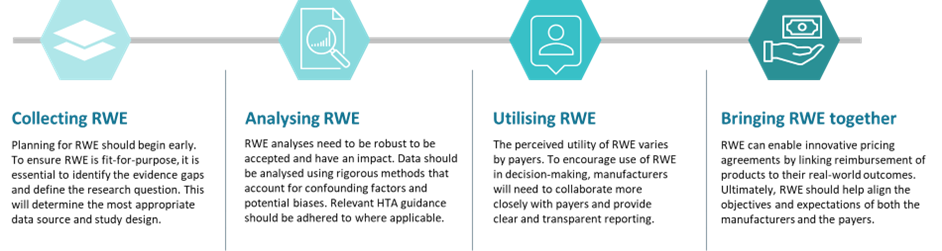
The feasibility of collecting RWE relies heavily on the availability of real-world data (RWD). However, most RWD are designed for billing or administrative purposes rather than for research. As such, collecting reliable RWE can be challenging because it often involves diverse patient populations, different treatment settings and methods, variations in data collection and analysis, potential biases and confounding factors, and difficulties in ensuring data quality and completeness. In Europe, the quality of RWD varies, requiring significant effort to curate the data to meet the requirements of the use case. Considering the challenges with RWD/RWE, the purpose of collecting RWD/RWE should be carefully considered to inform the research approach, otherwise it may lead to poor results. For instance, the long-term registry data did not support HTA reassessment for Myozyme in France, as it failed to show whether the drug slows the rate of progression, emphasising the need for RWE studies to be carefully designed to address key HTA considerations.20 As a first step, it is important to define the research question that the RWE intends to address, which will help identify the most appropriate data source and study design. Evidence gaps should be proactively identified, and RWE generation should be tailored accordingly. Planning for RWE should begin early in the product development process and be integrated as part of the global evidence generation strategy to support the product’s value and market access at launch.
The growing significance of RWE in HTA is apparent, as evidenced by the marked increase in its use in recent years. An analysis of HTA reports across 83 HTA bodies in 33 countries found that the use of RWE in HTA submission has risen sharply from 6% in 2011 to 39% in 2021.21 Incorporating RWE in HTA has shown to facilitate positive reimbursement decisions by providing external comparator data or additional data on intervention effectiveness, the validation of surrogate endpoints, and patient characteristics.21 For example, in the UK, RWE was used in a matching-adjusted indirect comparison (MAIC) demonstrating comparative effectiveness over its comparators (R-CHOP/R-CVP), leading to cost-effectiveness and contributing to a positive HTA outcome.22
RWE analyses need to be robust to be accepted by HTA bodies and have an impact. In Europe, formal guidance on the role of RWE to support HTA and reimbursement decision-making is emerging. In June 2022, the UK National Institute for Health and Care Excellence (NICE) published the most detailed HTA RWE framework to date. NICE recommends the following steps when analysing RWE: identify potential confounders using a systematic and transparent approach, use statistical methods to address confounding, consider the impact of information bias and address it appropriately, and use sensitivity and bias analysis to assess the robustness of results.23 The depth of HTA RWE guidance is still variable across markets, with most only providing high-level recommendations. Nevertheless, to optimise favourable reimbursement decision-making, manufacturers should ensure that RWE analyses are conducted rigorously to reduce the risk of bias and confounding and align these analyses with relevant HTA guidance and standards, where applicable.
Despite the growing importance of RWE, its acceptability and impact on HTA decision-making remains limited and highly variable among European markets.21 The perceived utility of RWE varies by payers. Those who have an ongoing responsibility for product assessment (e.g. as part of an MEA) are more likely to be receptive to using RWE compared to those payers whose mandate is focused solely on the initial assessment. In addition, many payers have reservations about incorporating RWE in their decision-making due to data quality, study design flaws, potential bias, and lack of meaningful endpoints.24 Payers should understand significant standards of scientific rigor are involved in the generation of RWE. Manufacturers must ensure clear and transparent RWE reporting, as well as closer collaboration with payers to realise the full potential of RWE.
While manufacturers recognise the need for RWE, greater strategic direction is needed to maximise its impact on access and commercial success. Generating robust, high-quality RWE is not sufficient alone and it should be harmonised with other pricing and reimbursement initiatives, namely innovative pricing agreements. RWE can help to define and measure the relevant outcomes that are aligned with the objectives and expectations of both the manufacturers and the payers. For example, in Spain, payments for Yescarta were linked to survival as collected through the Valtermed national registry system.25 The exact mechanisms and conditions for using RWE to implement OBAs remain difficult to define across products and markets. RWE beyond overall survival is still restrictive and there may not be a concerted administrative system in place to pool the data. To fulfill the potential of OBAs, manufacturers should work closely with payers to understand the type of RWE needed, and how it should be ultimately linked to inform access and pricing decision-making.
Conclusion
We have reached a global impasse where next-generation therapies present the potential to make a step change in patient outcomes, yet also carry a much higher price tag at a time when most nations endeavour to decrease healthcare expenditure. To maintain patient access in this environment, manufacturers and payers must work together as partners to co-create a hospitable, mutually beneficial, access environment across the three pillars of evidence generation, managed entry agreements, and real-world evidence.
While these appear as discrete functions, the three pillars are intrinsically linked, and overall product launch success will be dependent on synergistic execution. With this in mind, we recommend that manufacturers begin developing a strategy for how OBAs will be structured, and what RWE will be required to support, at the point of registrational trial design. By engaging payers in partnership at this early stage, the pharmaceutical industry can ensure that its launch strategy is aligned with prevailing market needs and tactically crafted to find the sweet spot between revenue and risk-sharing.
However, this is no ‘one size fits all’ solution. Markets across the EU have unique needs, depending on the method of assessment, economic environment, and policy changes, therefore the launch strategy must be tailored to the needs of individual payers. To achieve this product-market fit, manufacturers must engage with a diverse and fragmented cohort of decision-makers. This may seem outfacing but may be the only course of action to maintain patient access, revenue generation, and payer relationships in an uncertain economic climate.

Fill in the contact form and a member of your team will get back to you